Ago
15
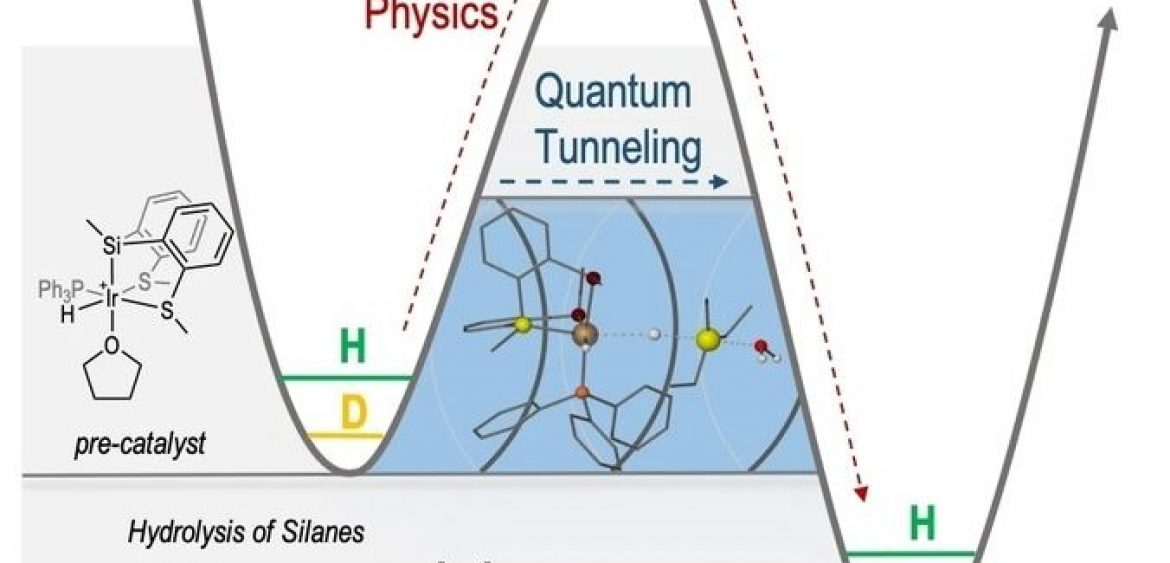
Hydrogen tunneling. Giant KIE!
Jun
27
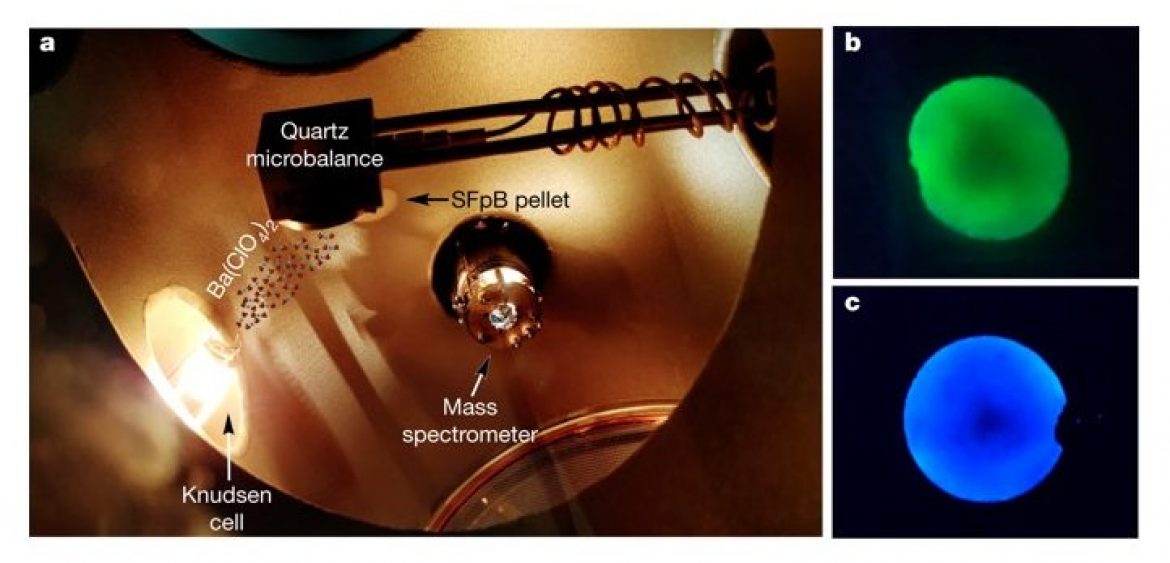
Fluorescent bicolour sensor for low-background neutrinoless double β decay experiments.
Jun
27
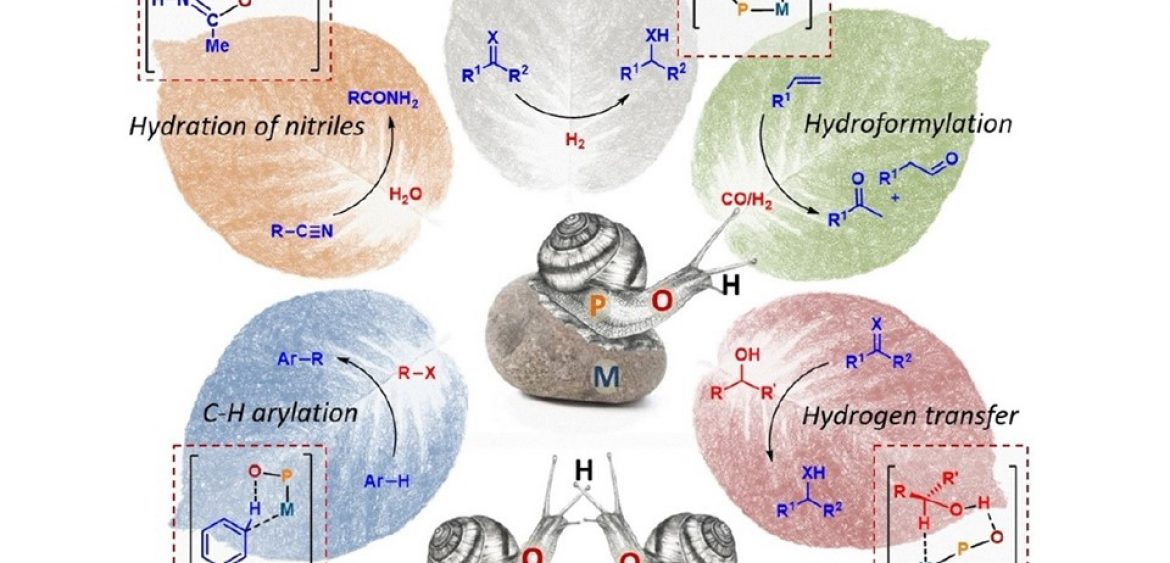
Secondary Phosphine Oxides: Bifunctional Ligands inCatalysis
May
21
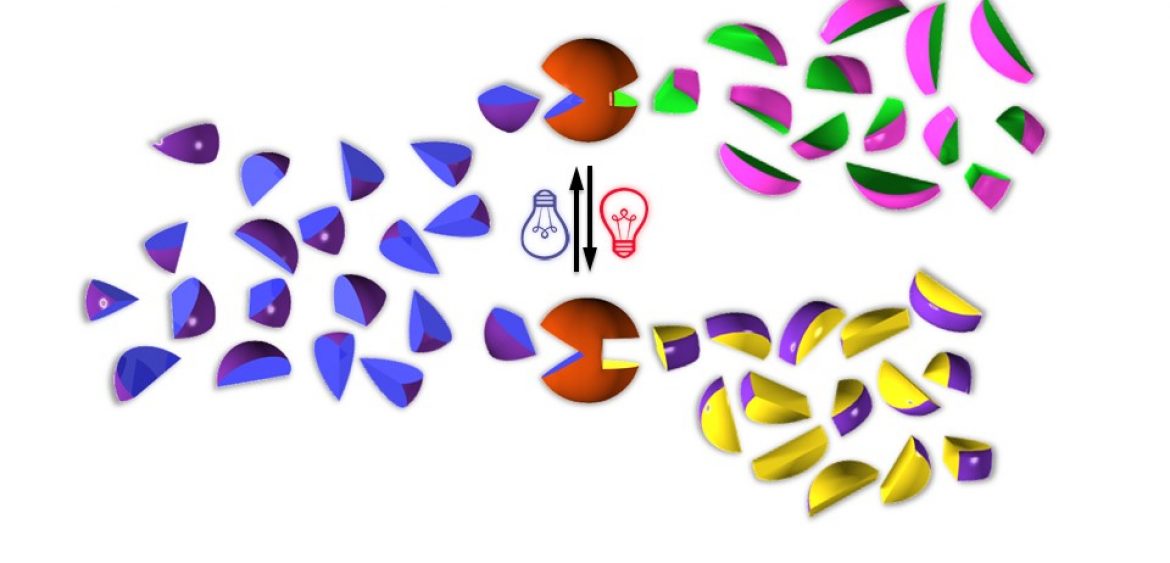
Photoswitchable catalysis using organometallic complexes
May
21
Proton-responsive Ruthenium(II) Catalysts for the Solvolysis of Ammonia-Borane
Oct
07
Revisiting the iridacycle-catalyzed hydrosilylation of enolizable imines… cover!
Oct
07
Light-driven water oxidation using hybrid photosensitizer-decorated Co3O4 nanoparticles
Últimos Posts
 PhD Day, Faculty of Chemistry UPV/EHU 2024 abril 20, 2024
PhD Day, Faculty of Chemistry UPV/EHU 2024 abril 20, 2024 PhD Ane I. Aranburu (31_3_2023) abril 16, 2023
PhD Ane I. Aranburu (31_3_2023) abril 16, 2023 Bachelor in Chemistry Graduation ceremony 2019/20 2020/21 agosto 16, 2022
Bachelor in Chemistry Graduation ceremony 2019/20 2020/21 agosto 16, 2022